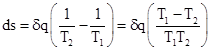|
Energy, Entropy, Enthalpy |
|
|
|
In the mechanical sense, work was originally defined in terms of lifting a weight to a certain height. The quantity of work was defined as the product of the weight and the height. This definition was then generalized, so that work was considered to be done whenever any kind of force is exerted through some distance. The quantity of work is the force multiplied by the distance. When two physical systems interact, one of them may do work on the other. We find it convenient to assign to each physical system a quantity called energy, with the same units as the units of work. Whenever a system does work on its surroundings, we say its energy has been reduced by the amount of work done, and whenever a system has work done on it (by some other system) we say its energy has been increased by that amount of work. By the law of action and re-action, all work that is done by one system is done on another system. It follows that the total amount of energy is conserved. (Notice that we haven’t established the absolute value of energy, we have merely discussed changes in the energy levels.) |
|
|
|
Classical thermodynamics is founded on two principles, both of which involve the concept of energy. The first principle asserts that energy is conserved, i.e., energy can neither be created nor destroyed, and the second principle asserts that the overall distribution of energy tends to become more uniform, never less uniform. These two principles are called the first and second laws of thermodynamics. |
|
|
|
In attempting to express the absolute energy content of a certain object in terms of familiar state variables, consider a stationary particle of mass m floating in empty space, and suppose we apply a force F to this particle over a distance Ds. By simple integration we know that an initially stationary object subjected to a constant acceleration a = F/m for a duration of time Δt will have traveled a distance |
|
|
|
|
|
|
|
The velocity v of the particle at the end of the acceleration is v = a Δt, so if we multiply both sides of the above equation by F we have |
|
|
|
|
|
|
|
Thus we might try to define the absolute energy of a macroscopic object as half the product of its mass times the square of its speed. However, if we take two identical lumps of clay and throw them together at high speed, the total system initially has energy according to our provisional definition, but after the collision it has none, because the lumps of clay stick together and the combined lump has zero speed. Therefore, this macroscopic definition of energy does not give a conserved quantity. This definition of energy is essentially equivalent to Leibniz’s vis viva, which literally translated means “living force”, but using the word “force” to signify what we today would call energy. When Samuel Clarke pointed out that this quantity is not conserved in such collisions, Leibniz replied |
|
|
|
The author [Clarke] objects that two soft or un-elastic bodies meeting together lose some of their energy. I answer no. ‘Tis true, their whole lose it with respect to their total motion, but their parts receive it, being shaken by the energy of the collision. And therefore that loss of [energy] is only in appearance. The energy is not destroyed, but scattered among the small parts. |
|
|
|
Here we recognize that in order for energy to be conserved we must consider not only the macroscopic kinetic energies of aggregate bodies, but also the microscopic kinetic energies of their constituent particles. The latter is usually regarded as the heat content of the aggregate body. Hence our concept of energy - if energy is to be conserved - must include not only the mechanical kinetic energies of aggregate bodies but also the internal heats of those bodies. |
|
|
|
However, even taking internal heat of massive objects into account, we can still find processes in which the quantity of energy seems not to be conserved. For example, a satellite in an elliptical orbit around a gravitating body moves more rapidly when it is near the gravitating body than when it is far from that body, so it’s kinetic energy changes significantly (while it’s internal heat content is not significantly altered). This shows that, to maintain the principle of energy conservation, we must include gravitational and other forms of potential in our definition of energy. (This relates to the original conception of work, which was based on raising objects in a gravitational field.) Likewise when we discover that material bodies can lose energy by emitting electromagnetic radiation, we must expand our definition of energy to include electromagnetic waves. This illustrates how we use the principle of energy conservation to define the concept of “energy”. We classify and quantify phenomena in whatever way is necessary to ensure that energy is conserved. (The great merit of the concept of energy is that the classifications and quantifications to which it leads are extremely useful, and provide a very economical and unified way of formulating physical laws.) |
|
|
|
Once we have developed our (provisional) concept of energy, we quickly discover that knowledge of the total quantity of energy in a given system is not sufficient to fully characterize that system. It’s also important to specify how the energy is distributed among the different parts of the systems. For example, consider a system consisting of two identical blocks of metal sitting next to each other in an isolated container. If the blocks have the same heat content they will have the same temperature, and the system will be in equilibrium and will not change its condition as time passes. However, if the same total amount of heat energy is distributed asymmetrically, so one block is hotter than the other, the system will not be in equilibrium. In this case, heat will tend to flow from the hot to the cold block, so the condition of the system will change as time passes. Eventually it will approach equilibrium, once enough heat has been transferred to equalize the temperatures. Thus in order to know how a system will change - and even to assess the potential for such change - we need to know not only the total amount of energy in the system, but also how that energy is distributed. |
|
|
|
The example of two blocks also illustrates the intuitive fact that the distribution of energy in an isolated system tends to become more uniform as time passes, never less uniform. This is essentially the second law of thermodynamics. Given two blocks in thermal equilibrium (i.e., at the same temperature), we do not expect heat to flow preferentially from one to the other such that one heats up and the other cools down. This would be like a stone rolling uphill. |
|
|
|
To quantify the tendency for energy to flow in such a way as to make the distribution more uniform, we need to quantify the notion of “uniformity”. Historically this was first done in terms of the macroscopic properties of gases, liquids, and solids. We seek a property, which we will call entropy, that is a measure of the uniformity of the distribution of energy, and we would like this property to be such that the entropy of a system is equal to the sum of the entropies of the individual parts of the system. For example, with our two metal blocks we would like to be able to assign values of entropy to each individual block, and then have the total entropy of the system equal to the sum of those two values. |
|
|
|
Let s1 and s2 denote the entropies of block 1 and block 2 respectively, and let T1 and T2 denote the temperatures of the blocks. If a small quantity δq of heat flows from block 1 to block 2, how do the entropies of the blocks change? Clearly the change in entropy can’t be just a multiple of the energy, because the total energy is always conserved, so entropy would always be conserved as well. We want entropy to increase as the uniformity of the energy distribution increases. To accurately represent uniformity, a given amount of heat energy ought to represent more entropy at low temperature than it does at high temperature. Therefore, it’s reasonable to weight the changes in energy by the inverse of the temperature. In other words, for a block of temperature T, we define the change in entropy ds resulting from the addition of a small amount of energy δq to be δq/T. (We assume δq is small enough that it does not significantly change the temperature of the block.) It follows that if a small amount of heat δq flows from block 1 to block 2, the entropy of block 1 will be decreased by δq/T1, and the entropy of block 2 will be increased by δq/T2, so the net change in entropy of the whole system is |
|
|
|
|
|
|
|
Thus the net change in entropy is positive if and only if the temperature T1 of the heat source is greater than the temperature T2 of the heat sink. |
|
|
|
Of course, we might have defined ds corresponding to the addition of a small amount of energy δq in some other way, such as –Tδq. Then the net change in entropy for our example would have been δq(T1 – T2), which again is positive if and only if T1 is greater than T2. However, defining entropy to be a negative value for the addition of heat seems rather incongruous. Also, recognizing that changes in the temperature of a macroscopic object are roughly proportional to changes in its energy, we could conceptually replace T with q (and δq with dq), so our two candidate expressions for the differential entropy are ds = dq/q and ds = –qdq. Notice that, up to an additive constant, the first implies s = log(q) whereas the second would imply s = –q2. When defined in the context of statistical thermodynamics we find that entropy is given by s = k log(W) where W signifies the number of microstates for the given macrostate. Thus, up to an exponent, we can roughly equate the heat content of an object with the number of microstates. |
|
|
|
Since entropy is a thermodynamic state property, its value depends only on the state of a system, not on the history of the system. Therefore, to determine the change in entropy of a system from one state to another, it is sufficient to evaluate the change for a reversible process between those two states; the change in entropy for any other process connecting the same two states will be the same as the change for the reversible process. (Note that this “conservatism” applies only to the entropy change between two states, not to the amount of heat flow associated with the process.) For large transfers of heat, such that the temperatures of the objects are significantly altered, we need only imagine a reversible “quasi-equilibrium” process of extracting heat from the hotter object and then another reversible process of adding heat to the colder object, and integrate the quantity δq/T in each case to give the total changes in entropy. |
|
|
|
Another useful state variable is enthalpy, defined as the sum of the internal energy and the product of pressure and volume. In other words, H = U + PV. The justification for defining this variable is really only a matter of convenience, because we often find that the sum U + PV occurs in thermodynamic equations. This isn’t surprising, because the work done by a quantity of gas depends on the product of pressure times volume. When a gas expands quasi-statically at constant pressure, the incremental work δW done on the boundary is PdV, so from the energy equation dU = δQ – δW we have δQ = dU + PdV. Noting that, at constant pressure, dH = dU + PdV, it follows that δQ = dH for this process. This explains why enthalpy is often a convenient state variable, especially in open systems. Obviously enthalpy has units of energy, but it doesn’t necessarily have a direct physical interpretation as a quantity of heat. In other words, enthalpy is not any specific form of energy, it is just a defined variable that often simplifies the calculations in the solution of practical thermodynamic problems. |
|
|